高リスク患者が参加しにくく、重い副作用が“控えめに見える”可能性
新薬の安全性は、基本的に**臨床試験(治験)**の結果をもとに判断されます。ところが近年、「治験に参加する患者さんが、実際に薬を使う患者像とズレていると、深刻な副作用(害)が過小評価されるのでは?」という問題が改めて注目されています。
全米経済研究所(NBER)のワーキングペーパーは、がん治療薬を例に、重い副作用が起きやすい患者ほど治験に参加しにくい傾向を示し、「治験データだけで害を見積もると控えめな数字になり得る」と報告しました。
✅ 先に結論:何が問題なのか
ポイントはシンプルです。
- 治験は参加条件が厳しい
- その結果、比較的“元気な患者”が集まりやすい
- すると **現実の患者(合併症が多い・虚弱など)**に起きる副作用が見えにくい
- **承認後に使われ始めてから「想定より重い副作用が多い」**となるリスクが上がる
つまり、治験は強力なエビデンスですが、外側(現実世界)への当てはまり=一般化可能性に課題が残ることがあります。
👥 なぜ高リスク患者は治験に参加しにくいのか
治験では通常、次のような要因で「高リスク患者」が除外・脱落しやすくなります。
📌 ① 参加条件(組み入れ基準・除外基準)が厳しい
- 合併症が多い人
- 肝腎機能が低い人
- 体力が落ちた人(フレイル)
- 併用薬が多い人
などは対象外になりやすい
📌 ② 手続き・通院負担が大きい
- 何度も病院に行く必要がある
- 検査が多い
- 同意や書類が煩雑
→ 体力・介助・交通手段の問題で参加しづらい
📌 ③ 医師側も「安全」を優先しがち
重い副作用が出やすい患者を試験に入れることは、倫理面・実務面でハードルが高くなりやすい、という現実もあります。
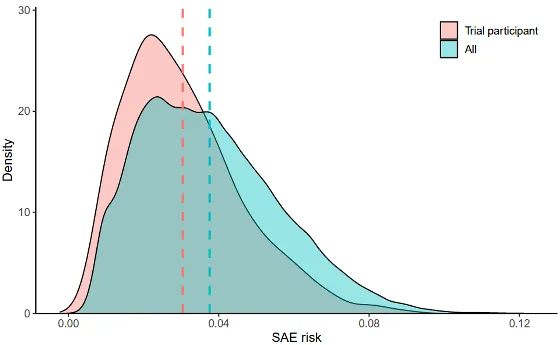
📉 NBER研究:治療開始後「重篤有害事象(SAE)での入院」が増える
研究チームは「薬の投与開始前後で、SAE(重篤な有害事象)による入院がどれくらい増えるか」を調べました。
🔎 SAEとは?
一般に、次のような深刻な副作用・出来事を含みます。
- 入院が必要
- 生命の危険
- 臓器不全
- 危険な感染症 など
📌 観察された規模感
- がん治療薬の投与開始後、SAEでの入院が増える傾向
- 平均的に 月あたり約2ポイント(例:患者100人なら月に2人増えるイメージ)
そして重要なのは「誰でも同じだけ増えるわけではない」点です。
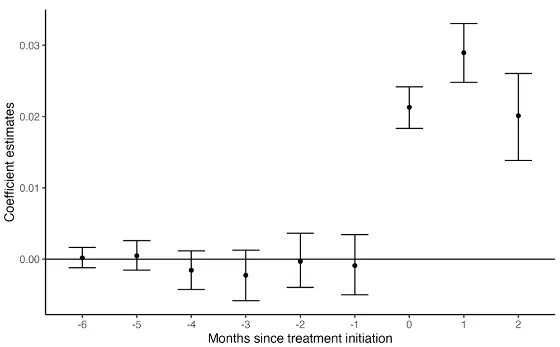
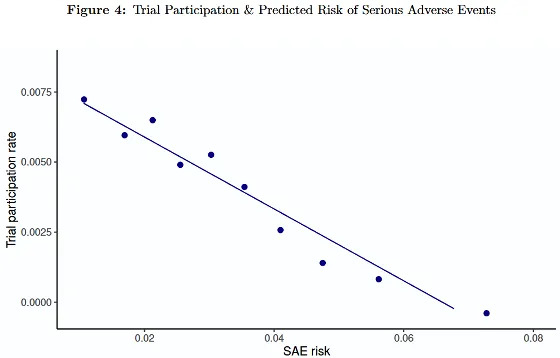
⚠️ リスクが高い人ほど“増え方”が大きい
研究では、合併症の多さや虚弱さなどから「SAE入院リスクが高い患者」を推定し比較しています。
- リスク分布の上位10%の患者は、下位10%に比べ
👉 治療開始後のSAE入院増加が 約2.5倍 と推定 - しかし上位10%の患者は、治験に参加する確率が
👉 約4分の1 だった
つまり、一番副作用が出やすい層が、治験データに入りにくい構造が示唆されます。
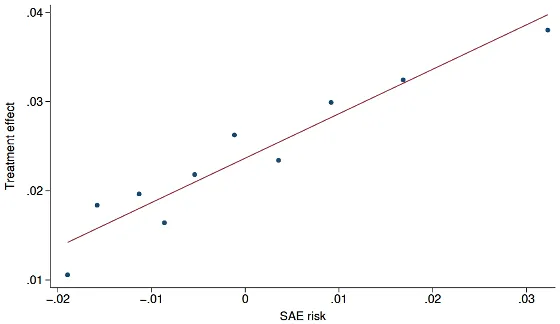
📊 「治験だけ」だと害が小さめに見える可能性(推定15%)
さらに研究チームは、この偏りが安全性評価に与える影響を推定しています。
- 現実の処方対象集団で見込まれる「薬が原因のSAE入院増加」は
👉 治験参加者だけを前提にした推定より15%大きくなる可能性 - 治療を1年続けると
👉 患者25人あたり1件くらい追加でSAE入院が起き得る、という試算
ここは医療者・患者双方にとって非常に重要で、
**「治験の安全性は嘘」ではなく「治験の安全性は“条件つきで見える”」**という話です。
🏛️ 規制当局はどう動いている?「代表性」と「多様性」の流れ
この問題は米国だけでなく国際的に議論されています。近年は、治験の参加者構成をより現実に近づける(代表性の向上)方向が強まっています。
🇺🇸 米国:Diversity Action Plan(多様性計画)
FDAは、臨床試験で過小代表になりがちな集団を含めるための**計画(Diversity Action Plan)**を求める枠組みを進めています(ドラフトガイダンス)。
※ただし、政策は政治状況などで揺れやすく、運用面の課題も指摘されています。
🌍 国際:ICH-GCPでも「不必要な除外を避ける」方向
ICHのGCP改訂(E6(R3))では、試験設計で不必要に特定集団を除外しないこと、参加者選定が「承認後に恩恵を受ける集団」を代表するように、という趣旨が明記されています。
🛰️ 「承認後」に埋める:リアルワールドデータと安全性監視
治験の弱点(代表性)を補う鍵として、承認後のデータ活用が重要になります。
🇺🇸 FDA:FAERS と Sentinel
- FAERS:副作用報告データベース(企業・医療者・患者から報告)
- Sentinel:大規模医療データを用いた安全性監視の国家システム
🇪🇺 EMA:PASS(市販後安全性試験)
EUでは、承認後に安全性情報を得る PASS の枠組みが整備され、当局委員会がプロトコルや結果を評価します。
🇯🇵 日本:PMDAとJADER、GPSP
日本でも市販後監視(PMS)や、
副作用報告DB JADER が運用されています。
💡 現場で起きやすい“誤解”を整理
ここ、医療ニュースで混乱しがちなポイントです。
❌「治験は信用できない」
→ 違います。
治験は因果推論に強い一方で、参加者の偏りが残り得ます。
✅「治験は“条件つきの安全性”を示す」
→ その通りです。
現実の患者群(高齢・多疾患・虚弱)に広がるほど、リスク推定は変わる可能性があります。
🧭 私たちができる“現実的”な対策
患者側・家族側の観点で、過度に不安にならずにできることもあります。
- 🧾 薬の「添付文書」「重要な副作用」を確認
- 🧑⚕️ 合併症・腎機能・肝機能・併用薬を医師に共有
- 📌 投与開始直後(副作用が出やすい時期)は、症状の変化をメモ
- 🗣️ 気になる症状は早めに相談(“様子見”しすぎない)
✅ まとめ 📝
- 治験は安全性評価の中心だが、参加者が「比較的健康」になりやすい
- 高リスク患者ほど治験に参加しにくく、重い副作用が見えにくい可能性
- NBER研究は、治験のみの推定が害を控えめに見積もるリスクを示唆
- 国際的に「代表性を高める」動き(ICH、FDAなど)が進行
- 承認後の監視(FAERS、Sentinel、PASS、JADER等)でギャップを埋めるのが重要
📚 参考・出典
- NBER Working Paper「Trials Avoid High Risk Patients and Underestimate Drug Harms」
- FDA:Diversity Action Plans(ドラフトガイダンス)
- ICH E6(R3) Good Clinical Practice(代表性・不必要な除外の回避)
- FDA:Sentinel Initiative(市販後安全性監視)
- FDA:FAERS(副作用報告DB)
- EMA:PASS(市販後安全性試験)
- PMDA:JADER利用規約・市販後監視/検査体制

